Notes from the Lab: T-Cell Engagers, the Next Stage in Cancer Immunotherapy?
Published:
01/27/2023

By Paul Bunk, Ph.D. candidate, Cold Springs Harbor Laboratory
In January 2022, tebentafusp-tebn, also known under the more easily pronounceable brand name Kimmtrak, was approved for the treatment of unresectable or metastatic uveal melanoma. This treatment is the first and, so far, only FDA-approved therapy for this rare kind of cancer. Tebentafusp-tebn (“tebe”) is also an example of a growing new class of immunotherapies, called bispecific T-cell engagers, which includes the leukemia drug blinatumomab (Blincyto). But what are T-cell engagers, how do they work, and why have they now proven to be effective in metastatic uveal melanoma, a cancer that seems to be resistant to other currently available immunotherapies?
How T-cells recognize cancer
To understand T-cell engagers, we first have to look at the body’s adaptive immune system. The adaptive immune system is tasked with recognizing and eliminating anything that can harm the body, including pathogens such as bacteria and viruses, as well as tumors. T-cells are part of the adaptive immune system and are tasked with directly killing cancerous or infected cells.
The adaptive immune system generally distinguishes between self and foreign—in other words, it differentiates between healthy cells of the individual and cancerous cells, or infected cells that pose a threat. T-cells distinguish self and foreign by recognizing fragments of proteins—we call these peptides—that are being presented to T-cells by MHC (major histocompatibility complex) molecules on the surface of cells. These MHC molecules tightly control the recognition of damaged or mutated peptides from infected or malignant cells by T-cells. In the case of infections, identification and destruction of infected cells by T-cells results in a rapid and strong immune response, due to the presence of peptides in viruses that are very different from those of healthy human cells.
In the case of cancer, identification and destruction of the malignant cells is less efficient, because human cancers have a limited number of peptides that T-cells can recognize as foreign as well as often lack the MHC molecules required to present those peptides to the T-cells. Some tumors harbor a large number of mutations, often due to high levels of DNA damage or lack of enzymes that repair DNA. This leads to a lot of cancer-reactive T-cells to enter the tumor and kill the tumor cells by recognizing its mutated peptides. This type of cancer is what we call immunologically ‘hot’. Other tumors have only a small number of mutations—what we refer to as a low mutational burden—and are called immunologically ‘cold’ due to the low number of T-cells in the tumor. This absence of T-cells is caused by the low mutational burden resulting in few mutated peptides that can be recognized by T-cells.
Besides the lack of immunogenic peptides in many tumors, T-cells often have to face other hurdles that limit their ability to enter the tumor and kill cancer cells. For example, even when a cancer has a high mutational burden and a lot of cancer-reactive T-cells, these T-cells often lose their ability to properly attack cancer cells inside the tumor. This immune ‘dysfunction’ is caused by a number of mechanisms, one of which includes tumors prompting the immune system to put ‘brakes’ (often called ‘checkpoints’) on T-cells. These ‘immune checkpoints’ are important in the protection of healthy, normal tissue from immune-mediated damage (which occurs in auto-immunity, including diseases such as lupus, rheumatoid arthritis, and multiple sclerosis). However, in the context of cancer, immune checkpoints are exploited by cancer cells to protect themselves from destruction by the immune system. Knowing that tumors utilize these ‘brakes’ and how they work inside the T-cell and in tumors led to Nobel prize-winning research and the development of a type of immunotherapy called immune checkpoint blockade. You might be familiar with several of the commonly used drugs for immune checkpoint blockade, including ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda), cemiplimab (Libtayo), atezolizumab (Tecentriq), avelumab (Bavencio), and durvalumab (Imfinzi). These drugs block the immune checkpoints and release the ‘brakes’ on the immune system, thereby reinvigorating T-cells so they can recognize mutated peptides within the tumor, resist the other suppressive factors within the tumor, and kill the cancer cells.
Coming back to our two types of cancers—tumors that have a high number of mutations and are immunologically ‘hot’ and tumors that have a low number of mutations and are immunologically ‘cold’: Immunologically ‘hot’ tumors have an existing T-cell response against the cancer cells that can be unleashed by immune checkpoint blockade treatment. Cutaneous melanoma, for example, harbors a high number of mutations; this immunologically ‘hot’ status explains why immune checkpoint blockade is effective for many patients with cutaneous melanoma.
Uveal melanoma, on the other hand, has a low mutational burden, meaning that there are not many mutated peptides for T-cells to recognize and react against, and as a consequence, not many T-cells are present within the tumor. Uveal melanoma is therefore referred to as immunologically ‘cold’, and it does not respond well to immune checkpoint blockade because there are not enough pre-existing T-cells to attack the cancer, even once the ‘brakes’ have been released. Furthermore, there are not enough mutated peptides on the cancer cells for the few T-cells that are present to recognize the cancer and initiate its destruction.
Outcomes for uveal melanoma patients have not improved in decades, but the tide may be shifting with the FDA approval of tebe, a new type of bispecific T-cell engager.
T-cell engagers prompt T-cells to kill cancer cells by bringing them close to each other
So how do T-cell engagers cause cancer cells to die? As described above, T-cells are part of our adaptive immune system and can directly kill cancer cells when they recognize those cells based on their mutated peptides that are presented by the MHC molecule on the malignant cells. This process is effective when mutational burden is high and there are a lot of mutated peptides to recognize, as with cutaneous melanoma. However, in tumors where mutational burden is low, T-cells simply do not recognize cancer cells as a threat because their lack of mutated peptides makes them appear like healthy cells.
Enter bispecific T-cell engagers. T-cell engagers are large molecules with two parts, one of which specifically binds to cancer cells, while the other specifically binds to and stimulates T-cells. T-cell engagers are given intravenously and then circulate in the bloodstream until they encounter a tumor cell displaying the antigen recognized by the T-cell engager. Once associated with the tumor cell, the T-cell binding portion of the engager can bind to any T-cell that is in the vicinity. This binding event keeps the T-cell in close proximity to the cancer cell and activates its biochemical machinery to undergo the steps required to kill the malignant cells.
On a molecular level, all this is possible because the two “binding” parts of the T-cell engager each bind to only a certain type of protein. The ‘cancer-specific’ part of the T-cell engager binds to proteins that are only present on tumor cells. We call these cancer-specific proteins tumor-associated antigens (TAAs). The ‘T-cell specific’ part of the T-cell engager binds to a protein that is expressed on all T-cells called CD3 (see Fig. 1).
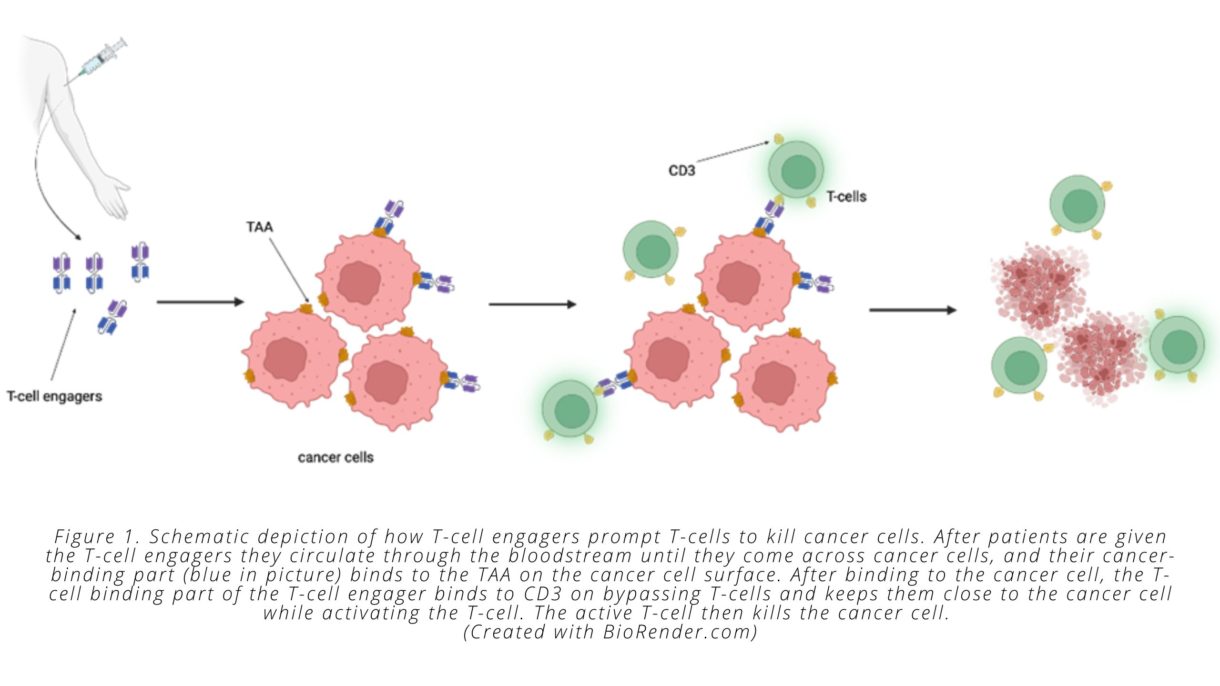
Importantly, T-cell engagers are able to overcome one of the challenges associated with immunologically ‘cold’ tumors. As noted above, these types of tumors do not contain a pre-existing cancer-specific immune response—meaning they don’t have a mass of dysfunctional cancer-specific T-cells present—that can be reinvigorated by immune checkpoint inhibitors. T-cell engagers circumvent this problem by prompting non-tumor specific T-cells to attack the cancer cells. This process means that T-cells can be redirected against cancer cells by the bispecific engager, regardless of whether the T-cell would otherwise have recognized a mutated cancer cell peptide, since the T-cell engager recognizes the TAA presented by the cancer cell and activates the T-cell through binding to CD3. Once T-cells have been activated and induced to kill cancer cells, they secrete molecules that recruit other T-cells, which causes more and more immune cells to infiltrate the tumor—another way that T-cell engagers solve the issues associated with ‘cold’ tumors.
To review so far: T-cells are immune cells that can kill cancer cells if the T-cells recognize mutated peptides on the cancer cell surface that differentiate the cancer cell from a healthy cell. But cancers that are immunologically ‘cold’ have few mutations and therefore a low number of mutated peptides for T-cells to recognize. Further, these cancers often have only few T-cells present inside the tumor, resulting in a low ratio of T-cells to tumor cells and poor T-cell killing of tumor. Bispecific T-cell engagers are drugs designed to solve these issues by redirecting T-cells to tumor antigens and by stimulating T-cells to kill tumor cells. The engager puts these two cells close together, activating the T-cells so that T-cells can kill the cancer cells. You can see how the name bispecific T-cell engager is an apt description of the drug, as it ‘specifically’ binds to two (‘bi’) cell types and ‘engages’ T-cells to kill cancer cells.
A new type of T-cell engager enters the clinic
The bispecific T-cell engagers described above—including blinatumomab, an engager directing T-cells to the CD19 protein on B cell leukemia cells—are engineered to recognize the malignant cells through an antibody-based structure, and they work because antibodies can specifically recognize and bind to proteins (the TAAs) on the surface of cancer cells.
Unfortunately, in cancer immunotherapy only 10% of all proteins are present on the surface of a cell and available for antibody-based T-cell engagers to recognize and bind to. That means that 90% of proteins—and therefore 90% of the markers that differentiate between cancer and healthy cells—will be present only on the inside of a cancer cell. A lot of cancers, then, would not respond to a bispecific T-cell engager, because the engager wouldn’t be able to identify a TAA on the surface of the cancer cell to distinguish it from healthy cells. But what if we could target the 90% of proteins inside of the cell as possible TAAs, so that T-cell engagers could identify the cancer cell and bind with it?
Here is where the innovation around tebe comes into play. Unlike the first T-cell engagers that employ two antibodies, tebe and other T-cell engagers currently under development use a T-cell receptor structure instead of antibody to recognize a peptide displayed by the MHC on the tumor cell surface. The other side, which is the part of the T-cell engager that binds to CD3 on T-cells, is an antibody that stimulates CD3, which is important for T-cell activation. Using T-cell receptors as part of a T-cell engager, therefore, allows the engager to bind to TAAs that wouldn’t be accessible to antibody-based T-cell engagers like blinatumomab. This difference is critical for treating cancers where there are no or only few TAAs found on the surface of the cell—such as uveal melanoma. Once T-cells are activated by a T-cell engager and start destroying cancer cells, signals are released, alerting nearby T-cells that cancerous cells are present, resulting in a cascade of T-cell-mediated cancer cell destruction.
The cancer-binding part of tebe targets a peptide derived from the TAA called gp100, a protein that is only present on the inside of melanocytes and melanoma cells. One caveat that comes with the use of T-cell receptor-based bispecific T-cell engagers is that the MHC molecule (which is required to recognize peptides on the surface of cancer cells) comes in many different forms that we inherit from our parents. A specific T-cell receptor can only recognize peptides displayed on selected MHC proteins. Tebe is targeted against gp100 presented by HLA-A*02:01, which is the most common MHC variant but is still present in only about half of all humans. In order to receive tebe for advanced uveal melanoma, patients will need to be tested for the presence of HLA-A*02:01, which is required for the drug to be active. Ongoing research is likely to develop other forms of bispecific engager that depend on different MHC types.
A first success in uveal melanoma
FDA approval of tebe was based on the results of a phase 3 clinical trial in which the drug showed statistically and clinically meaningful overall survival benefit for patients with unresectable or metastatic uveal melanoma (read the published article here). Uveal melanoma is an aggressive form of melanoma affecting the eye and eventually spreading to other parts of the body, almost always the liver, in about half of the patients. This tumor is very aggressive and has only rarely responded to any other form of therapy, making it an unmet clinical need with patients currently surviving on average only one year after metastasis is diagnosed. While this drug was the first to be approved for uveal melanoma, there are multiple other treatments for uveal melanoma still in clinical trials. These treatments include antibody-drug conjugates, combination therapies, cancer vaccines, and adoptive T-cell therapies. It remains to be seen how well these approaches fare in trials.
There are also a number of other T-cell receptor-based bispecific T-cell engagers currently under development, as other companies also believe in the potential of targeting tumor-specific peptides that are derived from proteins within cancer cells. This includes Immatics’ IMA401 and IMA402, which are being tested in a number of solid tumors such as non-small cell lung cancer and melanoma, and AbbVie’s ABBV-189 which is also being tested in various cancers. Despite this promising first approval of a T-cell receptor based bispecific T-cell engager, obstacles remain. As with other immunotherapies, tumors sometimes develop alternative mechanisms to resist the immune response or to escape from the immune response and grow back even after initial regression. Research is underway to develop better T-cell engagers and combinations with other therapies to increase the benefit of this novel immunotherapy and overcome resistance and escape from potent immunotherapy.

Paul Bunk is a Ph.D. Candidate at Cold Spring Harbor Laboratory, a New York-based research institute world-renowned in the fields of cancer biology, neuroscience, and plant biology. Paul works in the group of Semir Beyaz and focuses his research on the impact of metabolism on immune cells and their ability to fight cancer. With this research, the Beyaz Lab sits at the cutting edge of cancer immunology and hopes to help make better immunotherapies for the future.
Recent Posts

Jul. 20, 2026
More Than a Letter: A Year of Advocacy for Patients with Advanced Melanoma

Jul. 14, 2026
Finding Hope, Purpose, and Life Beyond Diagnosis

Jul. 10, 2026
Help Bring a New Treatment Option to Patients with Advanced Melanoma

Jul. 06, 2026
How Melanoma Changed Adam Harper’s Perspective on Life

Jun. 26, 2026